Weird multi-threaded behaviour of R/Bioconductor under Docker
As I was running some R code under Docker code recently, I noticed that processes that should be single threaded, were using all available threads. And this behaviour was different between R on a native Linux machine as compared to Docker.
A search of the forums found that this is due to the configuraiton of the BLAS system dependancy on those Docker images, which is set to use all available threads for matrix operations.
This configuration sounds like a good idea at first because dedicating more threads to a problem should speed up the execution. But realise that parallel processing incurs some overhead to coordinate the sub tasks and communicate the data to/from daughter threads. This means that you rarely achieve linear speedup the more threads you add.
Typically what happens is that parallelisation has a sweetspot where the first 5-10 threads provide some speed-up, but beyond that there is either no improvement in speed or that adding additional threads actually makes the code slower.
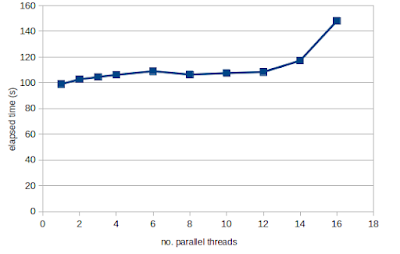
In the case of DESeq2 under Bioconductor 3.19 Docker, the default behaviour of using all threads is making the code a lot slower. And the effect of this is worse for more powerful multi-threaded computers. As per the forum post, this default option can be changed to a single thread using the following command from the RhpcBLASctl package:
`RhpcBLASctl::blas_set_num_threads(1) `
For example on our trusty 8-Core 16-Thread Threadripper 1990X system, DESeq2 of RNA-seq data consisting of 37 control and 37 cancer samples (code) takes 148 seconds with the default using all threads, but only 99 seconds using a single thread.
This also touches on a more general point that if you are writing packages, you shouldn't make assumptions about and use all available threads (see related prost from Jottr). There are many situations where this behaviour is undersirable:
- Shared systems where it would be rude to use 100% of available threads.
- Cloud systems and HPC where the number of threads available is different to nproc
- Where the number of available threads is really huge, such as modern server-grade systems with many threads (some have over 100 threads!)

