Using paired end read concordance to determine accuracy of RNA-seq pipelines STAR/featureCounts and Kallisto
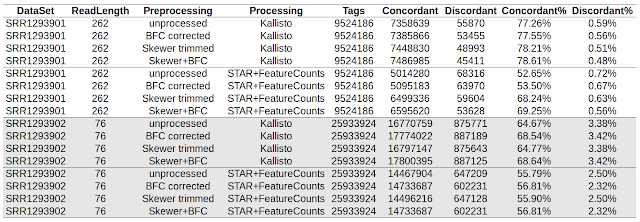
This post is a collection of code used to compare accuracy of Kallisto and STAR/featureCounts in my previous post . Download from SRA and convert to fastq format Check my older post. __________________________________________________________________ Run skewer to quality trim the data. $ cat run_skewer.sh #!/bin/bash for FQZ1 in *_1.fastq.gz ; do FQZ2=`echo $FQZ1 | sed 's/_1.fastq.gz/_2.fastq.gz/'` skewer -t 8 -q 20 $FQZ1 $FQZ2 done __________________________________________________________________ Run bfc to correct errors. $ cat run_bfc.sh #!/bin/bash for FQ in *.fastq.gz ; do OUT=`echo $FQ | sed 's/.fastq.gz$/_bfc.fastq.gz/'` bfc -t 8 $FQ | pigz > $OUT & done wait __________________________________________________________________ Run STAR aligner to align reads to human genome in single end mode. $ cat run_star.sh #!/bin/bash DIR=/refgenome_hsapiens/ GTF=refgenome_hsapiens/Homo_sapiens.GRCh38.78.gtf f...