Benchmarking an AMD Ryzen Threadripper 2990WX 64 thread system
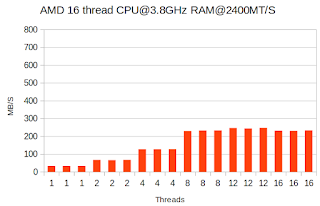
I recently received access to a new workstation built around the AMD Ryzen Threadripper 2990WX 32-Core Processor. The main use of the system will be to process genome sequencing data. It has 64 threads and the clock speed is 3.2GHz. It should get those data processing jobs done a lot quicker than my other system. Nevertheless I wanted to run some benchmarks to see how much faster it is as well as give it a stress test to see how well it can cope with high loads. This info could also be useful for comparisons in case degradation of the system in the future. Here are the specs of the 3 systems being benchmarked: AMD16: AMD® Ryzen threadripper 1900x 8-core processor - 16 threads@3.8GHz - 2.4GHz RAM Intel32: Intel® Xeon® CPU E5-2660 16-core processor - 32 threads@3.0GHz - 1.3GHz RAM AMD64: AMD Ryzen Threadripper 2990WX 32-Core Processor - 64 threads@3.2GHZ - 2.9GHz RAM The command being executed is a pbzip2 of a 4 GB file containing random data usin...