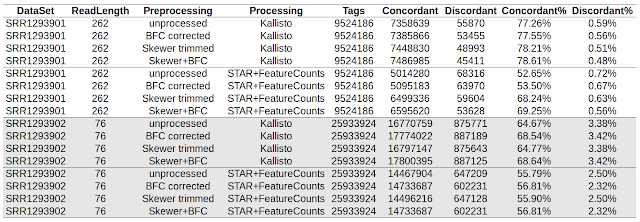
Introducing "Digital Expression Explorer"

RNA-seq has been a blessing for molecular biologists, not only does RNA-seq provide unbiased transcriptome wide expression analysis, it can be mined for a variety of other information like splicing, SNV identification, RNA editing, TSS usage, etc. As the cost of RNA-seq declines, more and more labs are using it hence more and more data is being deposited at databases such as SRA and GEO . But there is a growing problem. The problem is a lack of uniformity of processed data on GEO. Processed data has assorted reference genomes, gene annotation sets, accession numbers, software pipelines, statistical analyses and output formats, that in most cases makes comparison of two experiments hard if not impossible, let alone three or more experiments. This is a burden on researchers who want to quickly extract expression information from public RNA-seq data. Many researchers then resort to downloading the raw data in SRA format then processing it with QC/alignment/quantification/st...