Two subtle problems with over-representation analysis

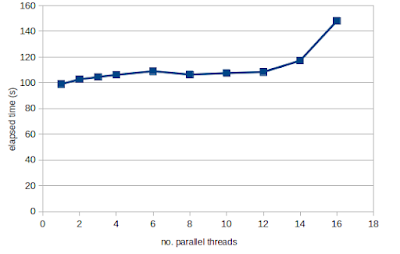
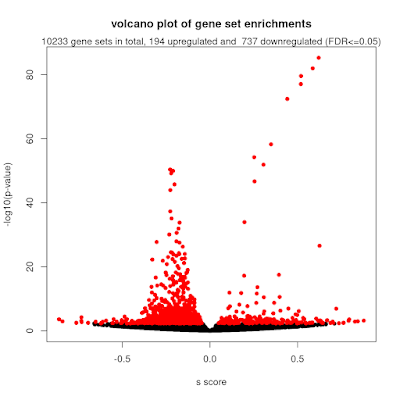
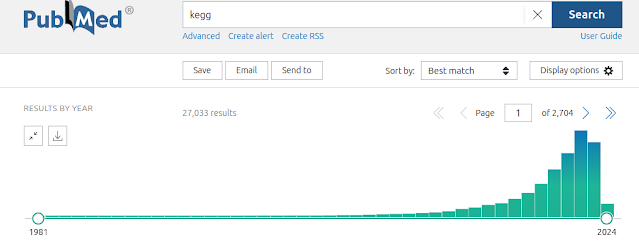
Over-representation analysis is a helpful and really frequently used technique for understanding trends from omics data and gene lists more generally. Just to demonstrate this, I tabulated the number of citations of some of the most popular web tools and packages, which reaches a massive 191k citations! But if you have used some of these tools, you will notice that they yield subtly different results. We were curiuous about that and ran a whole bunch of investigations into the internal workings of these tools, in particular clusterProfiler. We identified two subtle problems with clusterProfiler, which we unpack in detail in our new publication , but here I will give you a quick overview. Problem #1: The background problem The first one we call the “background problem,” because it involves the software eliminating large numbers of genes from the background list if they are not annotated as belonging to any category. This results in removing a large number of unannotated genes from the b...