Is paired end RNA-seq better than single-end for gene-wise gene expression analysis?
Something I've wondered about is whether for RNA-seq it's worth forking out the extra cost of sequencing both ends as opposed to single end.
To test this, I went back to a paired end data set present in GEO (GSE55123, 2x 36bp), cleaned the data with Skewer, then mapped the reads with STAR in either paired-end mode or single-end mode (using just read 1).
I then used featureCounts to quantify number of tags aligned to each gene. I excluded genes with fewer than 10 reads per sample on average. Then I ran edgeR at Degust to identify differentially expressed genes (DEGs@FDR<0.05). I used a shell script to quantify the overlap in DEGs. Then I ranked them based on the p-value from most up-regulated to most down-regulated and compared their positions in the rank.
Here's the result of the overlap analysis. You can see that PE fastq detected more genes but identified fewer DGEs than SE.
Detected in PE:15919
Detected in SE:15275
Detected in both:14750
Detected in either:16444
Detected in PE only:1169
Detected in SE only:525
Jaccard:89.6984%
DGE in PE:4651
DGE in SE:5755
DGE in both:4356
DGE in either:6050
DGE in PE only:295
DGE in SE only:1399
Jaccard:72%
Then I looked at the breakdown of where the 4651 PE DGEs ended up in the SE analysis:
DGE in PE and SE: 4356
PE DGEs not detected in SE: 138
PE DGEs detected but not significant in SE: 157
I then plotted the PE rank versus the SE rank for genes that were detected above threshold in both PE and SE. Rank is based on the sign of the fold change and the p-value. The result is near perfect.
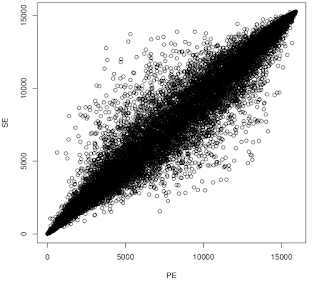
But I was curious to see whether the plot looked different if I included genes that were below the detection threshold in either SE or PE. The result is more consistent with the overlap analysis.

To test this, I went back to a paired end data set present in GEO (GSE55123, 2x 36bp), cleaned the data with Skewer, then mapped the reads with STAR in either paired-end mode or single-end mode (using just read 1).
I then used featureCounts to quantify number of tags aligned to each gene. I excluded genes with fewer than 10 reads per sample on average. Then I ran edgeR at Degust to identify differentially expressed genes (DEGs@FDR<0.05). I used a shell script to quantify the overlap in DEGs. Then I ranked them based on the p-value from most up-regulated to most down-regulated and compared their positions in the rank.
Here's the result of the overlap analysis. You can see that PE fastq detected more genes but identified fewer DGEs than SE.
Detected in PE:15919
Detected in SE:15275
Detected in both:14750
Detected in either:16444
Detected in PE only:1169
Detected in SE only:525
Jaccard:89.6984%
DGE in PE:4651
DGE in SE:5755
DGE in both:4356
DGE in either:6050
DGE in PE only:295
DGE in SE only:1399
Jaccard:72%
DGE in PE and SE: 4356
PE DGEs not detected in SE: 138
PE DGEs detected but not significant in SE: 157
But I was curious to see whether the plot looked different if I included genes that were below the detection threshold in either SE or PE. The result is more consistent with the overlap analysis.
In conclusion, these results show that going for the cheaper single end option loses about 10% of detected genes. It was quite surprising to see that single end data gave 23% more DGEs. I'm not sure whether these SE specific DGEs are actually real - we can't really know. Nevertheless, 93.6% of DGEs identified in PE were also identified in SE, so there aren't many DGEs that are missed in SE data. As the data set used here is relatively short length (36bp), as compared to current HiSeq standard (50bp), the choice to go single end will likely lose you only 5-10% of genes. This proportion of genes lost would be lower still at 100bp length. Unless you're interested in performing splicing analysis, my recommendation would be for 100bp SE RNA-seq with 40M reads per sample. I'd be interested in your own experiences with SE and PE RNA-seq.
