How accurate is Kallisto?
One of the most interesting developments in RNA-seq informatics in the past year or so is the evolution of so-called "lightweight" analysis. Instead of trying to map a whole read to the reference exons, it may be quicker and just as accurate to simply compare the k-mer content of the read and reference. If the read and reference share enough unique kmers, then the aligner can stop analysing that read and can move on the the next one. It turns out that the speed-up with this approach is huge, when compared with previous methods. Kallisto especially appears to have some speed advantages over competitors such as Sailfish, Salmon, eXpress, etc. For those interested in learning more, please refer to the Kallisto pre-print in arXiv, as well as the following blog posts.
For this post, I wanted to compare the accuracy of Kallisto with STAR/featureCounts (mapQ20 threshold), the pipeline that I'm currently using in most of my papers. I also want to know whether quality trimming with Skewer or error correction with BFC is also beneficial to accuracy (both default settings). The whole speed issue isn't a big deal for me as our department has more than enough computational resources available. I will use the number of concordant read pairs as a measure of true positives and discordant read pairs as a measure of false positives.
Kallisto installation was straight-forward, apart from the HDF5 dependency, which I had to guess the missing library. Ubuntu users should:
Here are the versions of the software, genome and data I used:
Kallisto uses a reference transcriptome sequence, which is different to STAR (genomic), so I made the transcriptome file contain both the transcript and gene ID in the header, so I can aggregate to get a read-gene relationship for concordance analysis. If Kallisto multi-mapping reads, then one was selected at random.
The data I used is from NCBI GEO (GSE57862) SRA (SRR1293901 & SRR1293902) and is useful because SRR1293901 is a 2x262 cycle run from Illumina MiSeq and SRR1293901 is a 2x76 cycle run from Illumina HiSeq 2000. The PLoS One paper that describes this dataset is also worth a look. The benefit of using this concordant read-pair approach is that it has all the quirks of real seq data that aren't accounted for by read simulators.
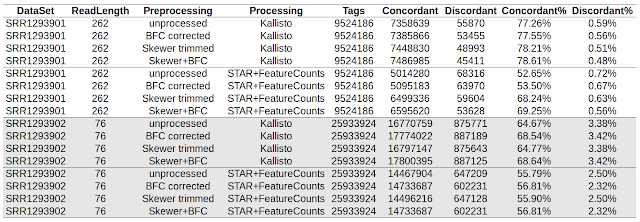
Firstly, you can see that in 262 nt data without pre-processing, Kallisto shows a very high concordance rate and low discordance compares to STAR/FeatureCounts. At 76 nt read lengths, Kallisto still shows a higher concordance rate, but discordance is also higher than STAR/FeatureCounts. Longer read lengths improve the concordance rates for Kallisto, while they actually decrease for STAR/FeatureCounts.
Skewer quality trimming was beneficial in all cases, but had a bigger impact on STAR/FeatureCounts than Kallisto pipeline. BFC error correction was also beneficial in all cases, but improved Kallisto pipeline to a greater degree.
In summary, Kallisto is not only a quick, but also accurate with longer reads and doesn't need huge RAM footprint like STAR does. The GEO dataset I used (GSE57862), will be a useful resource if you want to check concordance rates of other read lengths (ie:100 nt, 50 nt).
For this post, I wanted to compare the accuracy of Kallisto with STAR/featureCounts (mapQ20 threshold), the pipeline that I'm currently using in most of my papers. I also want to know whether quality trimming with Skewer or error correction with BFC is also beneficial to accuracy (both default settings). The whole speed issue isn't a big deal for me as our department has more than enough computational resources available. I will use the number of concordant read pairs as a measure of true positives and discordant read pairs as a measure of false positives.
Kallisto installation was straight-forward, apart from the HDF5 dependency, which I had to guess the missing library. Ubuntu users should:
apt-get install libhdf5-serial-devHere are the versions of the software, genome and data I used:
kallisto 0.42.2.1
STAR_2.4.0g1
featureCounts v1.4.2
bfc r181
skewer v0.1.122r
Ensembl Homo_sapiens.GRCh38.78 (cDNA, primary assembly, GTF)
Kallisto uses a reference transcriptome sequence, which is different to STAR (genomic), so I made the transcriptome file contain both the transcript and gene ID in the header, so I can aggregate to get a read-gene relationship for concordance analysis. If Kallisto multi-mapping reads, then one was selected at random.
The data I used is from NCBI GEO (GSE57862) SRA (SRR1293901 & SRR1293902) and is useful because SRR1293901 is a 2x262 cycle run from Illumina MiSeq and SRR1293901 is a 2x76 cycle run from Illumina HiSeq 2000. The PLoS One paper that describes this dataset is also worth a look. The benefit of using this concordant read-pair approach is that it has all the quirks of real seq data that aren't accounted for by read simulators.

Firstly, you can see that in 262 nt data without pre-processing, Kallisto shows a very high concordance rate and low discordance compares to STAR/FeatureCounts. At 76 nt read lengths, Kallisto still shows a higher concordance rate, but discordance is also higher than STAR/FeatureCounts. Longer read lengths improve the concordance rates for Kallisto, while they actually decrease for STAR/FeatureCounts.
Skewer quality trimming was beneficial in all cases, but had a bigger impact on STAR/FeatureCounts than Kallisto pipeline. BFC error correction was also beneficial in all cases, but improved Kallisto pipeline to a greater degree.
In summary, Kallisto is not only a quick, but also accurate with longer reads and doesn't need huge RAM footprint like STAR does. The GEO dataset I used (GSE57862), will be a useful resource if you want to check concordance rates of other read lengths (ie:100 nt, 50 nt).
